
We were happy to attend the latest EUnetHTA 21 stakeholder meeting dedicated to Health Technology Developers (HTD) on 13 July. As the EUnetHTA 21 Consortium will cease operations on 16 September 2023, the meeting started with a status update from EUnetHTA 21 before discussing the draft D5.4 deliverable “Joint Clinical Assessment (JCA) production timelines medicinal products”. This draft deliverable was one of two key topics on the agenda – the second being the draft D5.1 deliverable “Submission Dossier Template”. Here, we specifically highlight our learnings on the draft timelines.
While the timelines were not an official deliverable under the EUnetHTA 21 service contract, the group kindly drafted these to help stakeholders to prepare and better map required resources on a national and stakeholder level. The draft timelines are based on EUnetHTA JA3 experience, requirements in the Health Technology Assessment Regulation (HTAR) and the new EUnetHTA 21 deliverables. Notably, the revision of the pharma legislation has yet to be considered. The draft timelines are not scheduled for formal public consultation due to tight timelines for the overall scope of work being completed by the EUnetHTA 21 group before September. The draft could be revised by August, but only minor clarifications are expected.
The HTAR originally set two key JCA milestones for medicinal products:
- The dossier submission deadline is 45 days (at the latest) before an opinion is reached by the Committee for Medicinal Products for Human Use (CHMP);
- Endorsement of draft JCA reports must be submitted within 30 days after European Commission (EC) marketing authorization
Two possible scenarios are outlined. Scenario 1 for ‘new chemical entities (NCE)’ assessments within standard EMA procedures; and Scenario 2 for new indications of approved drugs (Type II variations) and accelerated assessments. The latter will require an earlier start date for JCA given its shorter EMA timelines, although EUnetHTA 21 is aware an accelerated assessment can revert back to a standard procedure.
Notably, the timelines for both scenarios are given in calendar days, rather than working days. It is hoped this will give some predictability and fairness between procedures, since the timeline cannot encompass all European national bank holidays and given that weekend days are part of the process in some of the steps. Stakeholders heard that several principles led to the general assumptions in the timeline.
These principles include:
- Predictable timelines for all parties involved; that is, a standardized process is needed
- HTAR requirements must be respected
- A balance is needed between having sufficient time for the anticipated workload versus the uncertainty of the regulatory process. There is considerable uncertainty, especially on a product’s final label, hence there is a balance needed between an early start without unnecessary risks
The general assumptions are that:
- Timelines are based on assumed, average standard regulatory process. Potential changes following the revision of the pharma legislation are not considered
- In order to meet HTAR milestones, the JCA must be conducted partly in parallel to the EMA assessment
- The timeline is based on average regulatory process, with an average duration of EMA clock stops, to be able to create a standard production process. EUnetHTA 21 notes that clock stops could be longer
- JCA production will not be interrupted before it is concluded
- However, JCA report will only be endorsed after final CHMP opinion to allow for a check of the approved indication compared to the claimed indication. Thus, in case of prolonged time between EMA submission and CHMP opinion, the Health Technology Assessment (HTA) Coordination Group (CG) review will be resumed once the CHMP opinion is available. There is no start and stop on other parts of that process.
For the two scenarios outlined, a different start date for the JCA is considered:
Scenario 1: NCE – start JCA approximately 2 months after the regulatory dossier submission to the EMA
Scenario 2: Type II variations or Accelerated Assessments – start JCA at the point of regulatory submission to the EMA
With the change in start date between Scenario 1 and 2, the rest of the timeline can be the same and the endorsement deadline can be the same, that is, 30 days after an EC decision.
The draft timeline only covers JCA with no relevant change to the claimed indication made by the HTD, as this will be the case in most procedures. Minor changes are expected to be managed with the Population, Intervention, Comparison and Outcomes (PICO) framework or PICO questions that will be asked. Major deviations can occur in practice as well, thereby affecting the content of the submission dossier and the JCA report. In such cases the advice from EUnetHTA 21 is to stop and re-start the JCA procedure, as the time available to address these changes is likely to be tight. EUnetHTA 21 recommends that a specific procedure should be developed that defines what constitutes a major deviation, who can decide on this matter, and whether the HTAR allows for this. Scenarios in which a change in indication is not reflected in the PICO could, for example, be considered a major deviation.
More from Lumanity on the EU HTA Regulation
Want to find out more about the EU HTA Regulation and how it will affect pharmaceutical companies bringing new products to market in Europe? Head over to Lumanity’s dedicated webpage where you can find all our updates on the Regulation so far, as well as key contacts and details of our training program.
Scenario I – New Chemical Entities (NCE)
Figure 1: The high-level timeline in comparison to the average, standard EMA process
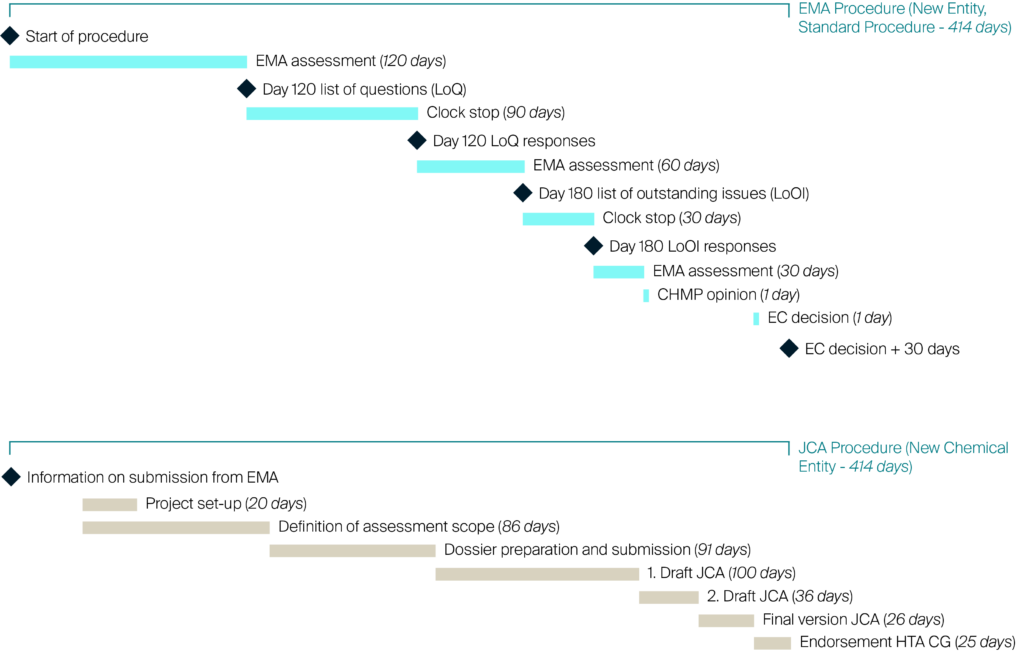
Table 1: JCA procedure for medicinal products with a hypothetical start date
| Procedure | Number of days | Start date | End date |
|---|---|---|---|
| JCA procedure (NCE – New Chemical Entity) | 414 | 01/01/23 | 04/02/24 |
| Information on submission from EMA | 0 | 01/01/23 | 01/01/23 |
| Project set-up | 20 | 13/02/23 | 04/03/23 |
| Identify assessors / co-assessors | 20 | 13/02/23 | 04/03/23 |
| HCP / patient survey | 20 | 13/02/23 | 04/03/23 |
| Definition of assessment scope | 86 | 13/03/23 | 07/05/23 |
| Information on drug / claimed indication to MS | 0 | 13/02/23 | 13/02/23 |
| Information from HCP / patient survey to MS | 0 | 05/03/23 | 05/03/23 |
| PICO survey | 14 | 19/03/23 | 01/04/23 |
| PICO consolidation | 14 | 02/04/23 | 16/04/23 |
| PICO validation | 20 | 17/04/23 | 06/05/23 |
| Information on PICO(s) to HTD* | 0 | 07/05/23 | 07/05/23 |
| Dossier preparation and submission | 91 | 08/05/23 | 04/08/23 |
| Dossier preparation by HTD* | 90 | 08/05/23 | 03/08/23 |
| Submission of dossier* | 0 | 04/08/23 | 04/08/23 |
| 1. Draft JCA | 100 | 05/08/23 | 09/11/23 |
| Check for formal completeness of dossier | 10 | 05/08/23 | 14/08/23 |
| Submission of requested additional content* | 10 | 15/08/23 | 25/08/23 |
| Preparation 1. Draft JCA by assessor / co-assessor | 100 | 05/08/23 | 09/11/23 |
| 2. Draft JCA | 36 | 10/11/23 | 15/12/23 |
| JCA SG review 1. Draft | 15 | 10/11/23 | 25/11/23 |
| Preparation 2. Draft JCA by assessor / co-assessor | 20 | 26/11/23 | 15/12/23 |
| Final version JCA | 26 | 16/12/23 | 10/01/24 |
| JCA SG review 2. Draft | 10 | 16/12/23 | 26/12/23 |
| Preparation final JCA by assessor / co-assessor | 15 | 27/12/23 | 10/01/24 |
| Endorsement HTA CG | 25 | 11/01/24 | 04/02/24 |
| HTA CG review | 8 | 11/01/24 | 19/01/24 |
| Medical editing | 5 | 11/01/24 | 16/01/24 |
| Fact check* | 5 | 11/01/24 | 16/01/24 |
| Incorporate comments, medical editing and fact check | 3 | 17/01/24 | 20/01/24 |
| Prepare version for HTA CG endorsement | 10 | 20/01/24 | 19/01/24 |
| HTA CG endorsement | 1 | 03/02/24 | 04/02/24 |
Notes: *timeframes relevant for the HTD
Source: Adapted from EUnetHTA 21 draft D5.4 deliverable
A high-level timeline, shown in Figure 1, illustrates the JCA procedure for Scenario 1, NCE. The figure shows how the draft timeline flows in comparison to the standard EMA process.
In earlier draft timelines shared by the EUnetHTA 21 group, HTDs would have just 30 calendar days from confirmed assessment scope (consolidated PICOs) to produce the submission dossier. Feedback from industry has pressed for more time, especially if more input on PICOs is expected. Given that feedback, the new timeline extends the dossier preparation time for HTDs to 90 calendar days from confirmed assessment scope (consolidated PICOs). The assumption is that manufacturers will have completed market research and will have already started preparations for the submission dossier. Importantly, the language of the dossier has not been decided yet.
The table above shows all the different steps and timings required to perform JCA. For the first draft JCA report, the timeline indicates 100 calendar days for preparation of the report by the assessor / co-assessor, which also includes the check for formal completeness of the submission dossier. Missing items may be flagged leading to requests for additional content to the HTD. Importantly, only 10 calendar days will be allocated for the manufacturer to supply the missing data. In parallel to CG review, the JCA report will go for medical editing and for a factual accuracy check. The factual accuracy check can be performed by the HTD who submitted the dossier; only 5 calendar days will be allocated for the manufacturer to complete fact checks on the final JCA report. The CG endorsed final report will be submitted to the EC within 30 days after EC marketing authorization.
Note, the EC have 10 working days from receipt of the endorsed reports to conclude that they do not comply with the procedural rules laid down in the HTAR so publication of the CG endorsed final report could be delayed beyond this 30 day post-marketing authorization window.
Scenario II – Type II variations and accelerated assessment
Scenario 2 outlines the assessment of new indications for approved drugs (Type II variations) and accelerated approvals. In these cases, the JCA process should be started earlier given the fast-track timelines adopted by the EMA for these assessments. The timeline runs parallel to the EMA timeline and the principles remain the same. The timeline starts at Day 0 of EMA submission. Time steps for this assessment remain unchanged.
Conclusion
The stakeholder meeting reviewed the draft timeline and confirmed it may be revised by August. EUnetHTA 21 recommended the following next steps:
- Timelines should be evaluated after a few JCA have been conducted under the HTAR, to assess the feasibility. Once a JCA is started, procedure and timelines should not be amended to ensure predictability for the HTD and assessors
- A process must be developed to deal with major deviations in the claimed indication
- A timeline calculation tool should be developed, so that timelines can be created easily and quickly, but also in a standard error-free way to ensure fairness
- Where possible, weekend days should be avoided
Overall, the timelines are challenging, as we were already anticipating. While we are happy to see that the timeline from confirmed assessment scope (consolidated PICOs) to dossier submission has been extended from 30 to 90 calendar days, timings are still tight, particularly for responding to missing data requests and factual accuracy checks post-submission. Therefore, we cannot stress the importance of early preparation enough. Lumanity has the expertise to support you in this endeavor.
Lumanity and independent HTA expert Neil Grubert have joined forces to deliver a cross-functional training program that raises awareness of the EU HTA Regulation’s impact. We are also providing support in responding to the consultation, in addition to planning for how the new Regulation will impact manufacturers’ portfolios, support evidence generation strategy, and develop a ‘business readiness plan’ to ensure your internal processes will align with the requirements of the Regulation.
Contact us to find out how we can be your strategic partner and help you to navigate a path through this evolving landscape.


